FDA Sends Record-Low Warning Letters To Device-Makers In 2017 As Agency Takes More Personalized Compliance, Enforcement Tack

Executive Summary
US FDA's ongoing compliance and enforcement outreach to industry – including holding regulatory meetings with and/or sending untitled letters to troublesome manufacturers – appears to be a contributing factor for why the agency sent an historically low number of quality-related warning letters to device-makers last year.
But officials in FDA's Center for Devices and Radiological Health stressed in an interview with Medtech Insight that there's no specific reason that a mere 35 warning letters were issued to firms in 2017, a decrease of 39% from 2016, when FDA wrote 57 missives. (See Figure 1.)
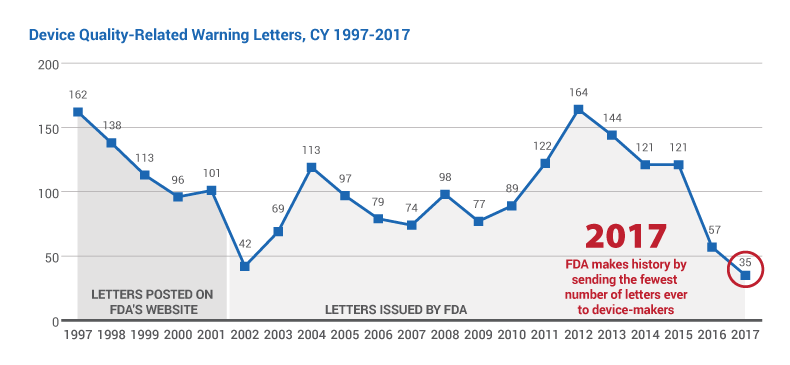
The agency's tabulation of 35 warning letters last calendar year – the lowest number recorded since the Quality System Regulation came into force in 1996 – tracks closely with its fiscal year 2017 count of 33 letters, which were handed out between Oct. 1, 2016, and Sept. 30, 2017.
FDA considers quality-related letters to be those that only include an alleged violation of the QSR. In its count, the agency does not include letters that solely pertain to the Medical Device Reporting (MDR) regulation (21 CFR, Part 803) or the Corrections and Removals regulation (21 CFR, Part 806), or pre-market activities.
"There's not necessarily a single factor that we would point to for any reduction or increase in the numbers from one year to the next," said Sean Boyd, deputy director for regulatory affairs within CDRH's Office of Compliance.
"Warning letters are just one of kind of many compliance and enforcement tools that we use to address issues with regulated industry. We also conduct regulatory meetings and issue untitled letters, both of which are examples of interactions that are not necessarily publicly displayed, but they communicate quality system deficiencies or issues with firms that have been inspected," he said. "Many of these issues are resolved using approaches like those, which we believe is one reason for the decrease" in warning letters.
Last year's all-time low letter count comes only five years after the agency sent the most letters ever to industry; it issued 164 in 2012. (Also see "Record Number Of Warning Letters Issued In 2012; Complaint Handling Troubles Significant" - Medtech Insight, 14 Mar, 2013.)
Letters Posted Online
For its part, Medtech Insight counted 42 quality-related warning letters posted on FDA's website in CY 2017. Check out our Warning Letter Data Tracker here.
Boyd noted that it's difficult to gain a full picture of industry's quality systems troubles and hot spots – and how they're being addressed by manufacturers – by only reading warning letters.
Rather, "what's important to us at FDA is that we're conveying inspection observations and findings to the firms using any means necessary," he said. Whether it's a warning letter, untitled letter, regulatory meeting or some other type of back-and-forth communication with device-makers, "we believe there are sufficient mechanisms in place to ensure that firms understand what the issues are and what's expected to address them."
Although Boyd says industry activities around the Case for Quality – a popular initiative run jointly by FDA and the Medical Device Innovation Consortium (MDIC) that urges firms to make quality an organization-wide concern – "isn't necessarily a reason for the decline in the issuance of warning letters," he did point out that the agency's work around benefit/risk concepts probably had some effect.
"Benefit/risk does impact how we evaluate inspectional findings and how we interact with industry to respond to those inspectional findings, whether it’s using a compliance and enforcement approach, or whether it’s taking a different interactive approach with a particular firm," Boyd explained.
A 2016 guidance document from FDA outlines a broad framework for considering benefit/risk factors in medical device availability, compliance and enforcement decisions. The guidance specifically targets post-market determinations and asks firms to consider submitting relevant benefit/risk information to the agency. (Also see "'A Sea Change': Device Center Compliance Chief Touts US FDA's Benefit-Risk Concepts – But Will Manufacturers Buy In?" - Medtech Insight, 7 Aug, 2017.)
"That document was published to align both industry and FDA on benefit/risk and other factors to consider when looking at compliance activities, and internally CDRH has considered those factors in determining whether to issue a warning letter, whether to issue an untitled letter, or take some other approach to communicate issues and resolve those with firms," Boyd said.
"There isn’t a quota. There’s not a specific number of warning letters that we aim for. We issue the letters that we think are appropriate," FDA's Carl Fischer says.
Of the 33 warning letters sent to firms in fiscal year 2017, 22 went to domestic device-makers and 11 went to overseas ones. (See Figure 2, "Quality-Related Warning Letters: Domestic Vs. Foreign, FY 2010-2017.")
"If you look at the total number of device facilities and you look at the number of inspections – and consider that we also inspect sites based on risk – I think you would expect to see some year-to-year variability in the number of warning letters," said Carl Fischer, senior advisor in CDRH's compliance office. "There isn’t a quota. There’s not a specific number of warning letters that we aim for. We issue the letters that we think are appropriate."
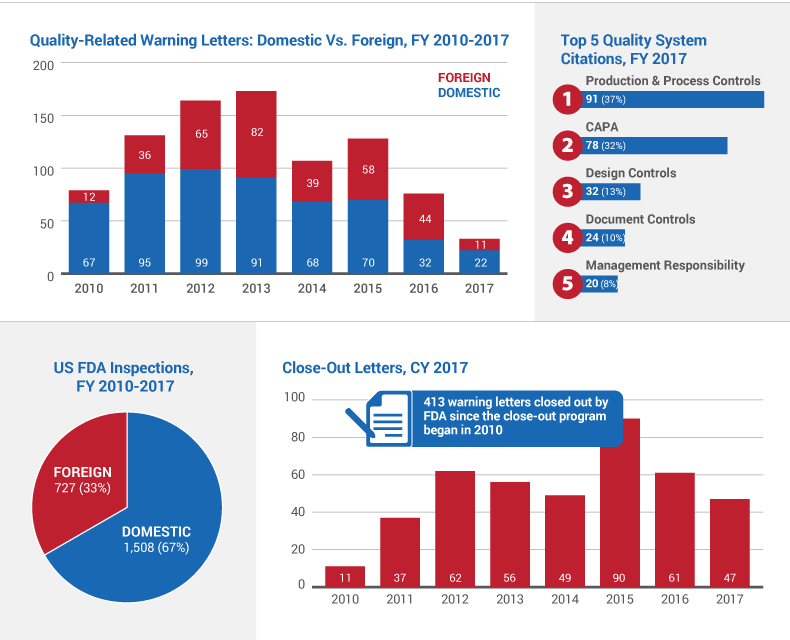
P&PC, CAPA Top Observations
When it comes to specific inspectional observations, device manufacturers had the most trouble with production and process controls; P&PC problems were noted in 37% of quality-related warning letters in FY 2017. That was followed closely by corrective and preventive action (CAPA) citations, which were found in 32% of missives.
That CAPA was cited on many warning letters isn't a surprise given that corrective and preventive action is always reviewed during an abbreviated Level 1 FDA inspection. Such an audit allows investigators to examine CAPA plus one other quality system subsystem, including production and process controls, or design controls.
"When an investigator looks at a CAPA, usually they look at a CAPA that is impactful to the product and can impact the product’s quality. It's natural, then, when an investigator sees a CAPA, to go to production and process controls because frequently the CAPA was initiated because of an issue, or they’re somehow connected," Fischer said, explaining how CAPA and P&PC activities – and therefore violations – can be intertwined.
"It’s likely that when an investigator looks at CAPAs, and they look at a sample of CAPAs and look at samples of complaints, that they then trace that back to production and process controls," he added.
Activities related to design controls (found in 13% of letters), document controls (10%) and management responsibility (8%) also were problematic for device-makers last year. (See Figure 2, "Top 5 Quality System Citations, FY 2017.")
Expert Process Validation Advice
When it comes to production and process controls, manufacturers often fall down when conducting process validation activities under QSR Sec. 820.75. Check out these recent Medtech Insight Compliance Corner articles and Compliance 360° podcast on the topic for tips and advice from top industry insiders:
Close-Out Letters Continue Downward Slide
Meanwhile, FDA sent 47 letters to firms under its warning letter close-out program last year, according to a Medtech Insight count of close-out missives the agency posted online in CY 2017.
Under that program, the agency sends a letter to a company and posts it to its website when concerns outlined in a warning letter have been addressed. Such correspondence becomes part of a manufacturer's inspectional history.
The 47 close-out letters given to manufacturers in 2017 is a 23% decrease from 2016, when 61 close-outs were issued; it brings the total number of close-outs to 413 since FDA launched the program in 2010. It also continues a year-on-year downward trend for the number of close-outs issued. (See Figure 2, "Close-Out Letters, CY 2017.")
"Warning letter close-outs are dependent on the number of warning letters issued, so it makes sense if there is a decline in the number of close-outs that [Medtech Insight] counted, commensurate with or in parallel to the decline in the number of warning letters issued," FDA's Boyd said. "Close-outs are dependent on interactions between industry and FDA, and industry’s follow-through on corrective action plans to adequately address those inspectional findings.
"The time to resolve those issues vary depending on the number and complexity of the issues, and the timeframe within which firms can remedy the observations, or the violations that are observed," he added, noting that the agency "doesn't have any plans to change the close-out process. We intend to continue to implement the program."
FDA Might Apply 'Least Burdensome' Concepts To Warning Letter, Close-Out Processes
Moving forward, however, FDA is looking to identify opportunities to improve the close-out letter process – and even its warning letter process – by applying "least burdensome" principles.
In FDA device regulation, least burdensome signifies an approach that seeks to minimize unnecessary steps and requests on the part of regulators or industry. The concept has been around for more than two decades, but 2016's 21st Century Cures Act required the agency to put it to work in some new ways, including provisions that FDA conduct more staff training focused on least burdensome principles, undergo an ombudsman audit of its practices, and ensure that the least burdensome approach is applied to some specific circumstances during pre-market review. (Also see "'Least Burdensome' Supersized? Draft Guidance Outlines An Expanded View Of The US FDA Concept" - Medtech Insight, 14 Dec, 2017.)
But FDA went one step further in a draft least burdensome guidance document it released in December, noting that the approach should extend to the agency's post-market activities.
Least burdensome "could apply to all our compliance engagements, pre-market and post-market," FDA's Sean Boyd says.
"Least burdensome ensures that we collect the minimum amount of information needed for the decision or issue at hand, using the most efficient means available to resolve those questions or issues, which is provided at the right time either in the lifecycle of the device or in the particular review that we’re performing," Boyd said.
"In the case of inspectional interactions, least burdensome approaches could streamline the way we interact and follow-up to an inspection, or when we’re requesting additional information from industry to evaluate and ensure that those findings are remedied, which could impact, for example, warning letter close-outs," he said.
Least burdensome "could apply to all our compliance engagements, pre-market and post-market," Boyd added.
From the editors of The Gray Sheet