FDA Quietly Sunsetting Summary Reporting Program For Adverse Events, Readies Public Release Of Millions Of Pre-2017 Summarized MDR Reports

Executive Summary
The US FDA expects to turn the lights out on its 22-year-old Alternative Summary Reporting Program for adverse events at the end of May, the agency tells Medtech Insight. But it's the FDA's upcoming online release of millions of summarized Medical Device Reports sent to the agency between 1998 and 2017 that could have the tongues of industry – and the public – wagging.
When the US Food and Drug Administration announced it was shutting down its Alternative Summary Reporting Program for adverse events, it did so with little fanfare.
The agency quietly made the announcement on its Medical Device Reporting webpage on 2 May. On that same day, news of the program's imminent demise was buried in paragraphs nine and 10 of a rather lengthy joint press statement on breast implant safety by FDA Principal Deputy Commissioner Amy Abernathy and Center for Devices and Radiological Health Director Jeff Shuren. (Also see "Breast Implant-Makers Now Required To File Individual Reports On Each Adverse Event, FDA Says" - Medtech Insight, 3 May, 2019.)
"In an effort to promote greater public transparency, the FDA has ended all summary reporting of breast implant Medical Device Reports and has notified breast implant manufacturers of this decision," Abernathy and Shuren wrote. "This is part of a larger effort to end the Alternative Summary Reporting Program for all medical devices, which we intend to complete in the coming weeks."
Since then, the agency hasn't said much more about its plan to ax the program – and "in the coming weeks" is a squishy timeframe to place on putting an end to a program that took in millions of summarized adverse event reports since January 1998.
But on 15 May, FDA spokesperson Deborah Kotz confirmed to Medtech Insight that the agency is working with device-makers to end the program for good by the end of the month.
The FDA's Alternative Summary Reporting Program kept millions of safety reports hidden from public view.
The ASR Program allows firms to submit abbreviated reports in a summarized, line-item format; ASR exemptions are currently in place for only three product codes. The program was established in 1997 in an effort by the agency to review adverse events more efficiently for well-established risks. (Also see "MDR summary reporting expansion planned by FDA; program begins with about 30 firms." - Medtech Insight, 29 Sep, 1997.)
While the agency's plan to end the program appeared to come out of the blue, Kotz said the FDA made the decision to slowly wind it down last year "in light of the statutory mandate for FDA to create a VMSR Program."
VMSR is the agency's Voluntary Malfunction Summary Reporting Program, launched in August 2018. It addresses goals outlined in a 2016 MDUFA IV commitment letter that directed the FDA to allow manufacturers of a majority of devices to report adverse events quarterly in a summarized, line-item fashion. (Also see "Makers Of An Array Of Devices Can Now Report Malfunctions In Summaries To FDA. Did Your Product Make The Cut?" - Medtech Insight, 17 Aug, 2018.)
Firms do not have to apply to the agency to participate in the VMSR Program; instead, they can "self-elect" to take part, the FDA says. Products that fall under the program's purview are low-risk class I devices, and class II products that aren't permanently implantable, life-supporting or life-sustaining.
Adverse events that result in a death or serious injury cannot be reported under the VMSR Program. Such events must still be reported to FDA within 30 days (or within five days if a problem with a device is particularly egregious).
The VMSR Program "is important to patient safety because it frees up [agency] resources to better focus on addressing the highest risks, such as deaths and serious injuries associated with medical devices," Kotz said. "This program also enables us to more efficiently detect potential safety issues and identify trends, thereby enabling FDA to be more effective in its device safety oversight."
Reports used in the VMSR Program – unlike those for the ASR Program – include the number of malfunctions described and are made publicly available via FDA's Manufacturer and User Device Experience (MAUDE) database, where all MDRs are stored.
The sunsetting ASR Program "only applied to specific malfunctions and serious injuries, not to deaths," Kotz said. "FDA carefully reviewed and considered all such reports, but they were not made publicly available because, prior to 2017, ASRs were not submitted in a format compatible with the public databases.
"The agency recognized the public interest in this information and modified the conditions of the ASR Program in 2017 to require submission of a 'companion' report on the official mandatory reporting form so that information collected through the ASR Program would be visible publicly," she added. "Each companion report includes the total number of events that are being summarized for that quarter" and is publicly available and searchable in MAUDE.
"FDA will communicate with the public when we have completed sunsetting the ASR Program and have made all ASR data publicly available." – Deborah Kotz
In what Kotz calls "an effort to promote greater public transparency," the FDA says on its MDR webpage that it will publicly release all summary reports filed with the agency before 2017, also "in the coming weeks."
The ASR Program was the center of controversy in early March after an investigative report was published by Kaiser Health News spotlighting safety issues with surgical staplers, which the report says have stayed hidden from public view because of the program. (Also see "FDA Eyes Upclassification, Labeling Guidance To Address Stapler Risks" - Medtech Insight, 8 Mar, 2019.)
The ASR Program was expanded to include surgical staplers in January 2001. (Also see "CDRH To Start New Year By Expanding Alternative Summary Reporting" - Medtech Insight, 1 Jan, 2001.)
While a key goal of the ASR Program was to minimize duplicative reports that can overwhelm the agency's surveillance activities, it also had the effect of keeping millions of safety reports in the shadows.
Millions Of Reports To Be Made Public
The FDA's plan to release all pre-2017 summary adverse event reports might prove tricky for the agency given the sheer volume of the reports.
The agency said in 1997 that the ASR Program would eliminate roughly 50,000 individual reports each year.
But, as it turns out, the program eliminated many, many more than that. Between 2001 and 2010, summary reports routinely outnumbered adverse events sent to the FDA individually on full MedWatch reporting forms. (See chart below.)
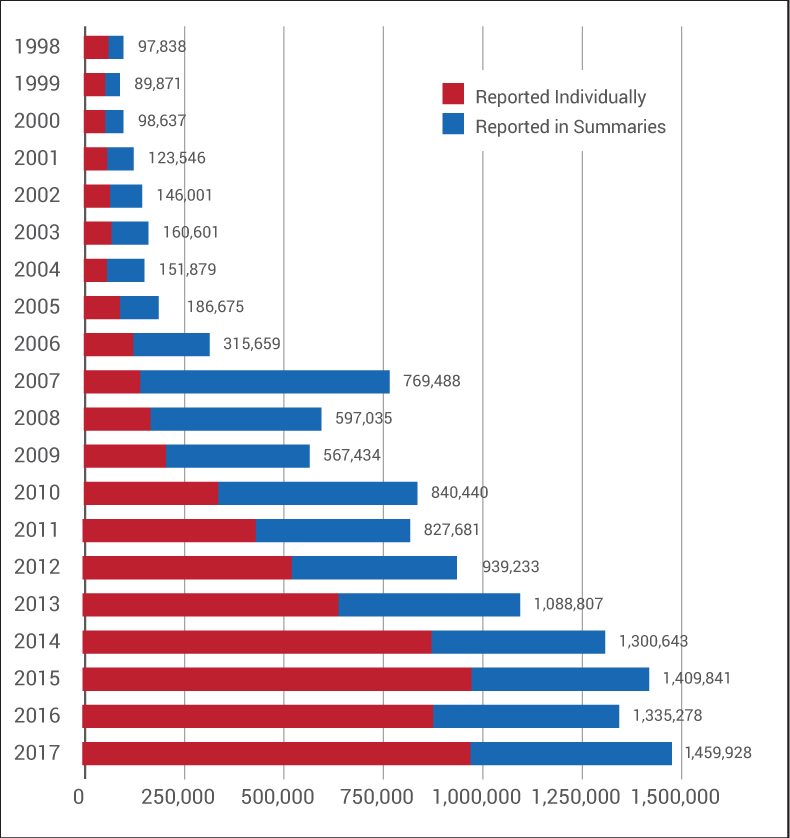
In 2006, an FDA official told Medtech Insight that summary reports made up 40% to 50% of the agency's MAUDE database. (Also see "As Industry Debates Postmarket Issues, FDA Says Firms Underreport MDRs" - Medtech Insight, 15 May, 2006.)
And the number of summary reports continued to balloon from there. Two years stand out in particular: 2007, which saw the largest number of summary reports ever sent to the agency in a given year (628,083), and 2010, when there were more than half a million summary reports filed.
Further, in 2015, 2016 and 2017, the FDA took in nearly 1.5 million summary MDRs under the ASR Program. (Medtech Insight requested from the FDA adverse event numbers for 2018, but Kotz said the agency is "still analyzing" last year's numbers and won't have that data ready "for a few weeks.")
All of that adds up to one giant data dump on the public by the FDA, coming soon.
The agency "will communicate with the public when we have completed sunsetting the ASR Program and have made all ASR data publicly available," Kotz promised.