Balancing Innovation And Safety – The US and Australian Medtech Regulatory Systems Compared
Executive Summary
Effective medical device regulation supports both safety and innovation needs. This article assesses how well the US FDA and the Australian TGA achieve this balance.
Health care products manufacturers face increasingly demanding regulatory landscapes to ensure that products entering the market are safe and effective. There is a growing focus on globalized regulatory approaches, the Medical Device Single Audit Program (MDSAP) being a good example. Global approaches necessitate the need to gain an appreciation of the differences in the regulatory strategies of different global regions.
For medical device start-ups working on innovative products, registration is a significant milestone. Start-ups may choose to register in a market where the regulator is known to be familiar with novel products. Start ups must weigh up the advantages and disadvantages of the various systems available to be able to make informed choices about their own regulatory strategies.
Systems That Encourage Innovation
The effectiveness of regulation can be measured in how a product’s quality, safety and performance is assessed before it enters the market, and during its lifecycle.
How regulatory authorities encourage novel solutions, and assist businesses in bringing innovations to market, are pivotal considerations for manufacturers developing market entry strategies.
It is evident that both the the TGA and FDA are reforming to encourage innovation in medical devices in their respective systems.
Overall, the Australian Therapeutic Goods Administration (TGA)'s regulatory approach is aligned with that of the rest of the world, which is one of reducing the regulatory burden and offering an effective, risk-based and lifecycle approach to regulation. The US Food and Drug Administration (FDA)’s 510(k) substantial equivalence route does not encourage innovation, and is a less evidenced-based method of regulation.
This article compares the FDA and TGA, and makes an assessment as to which system better supports the effective regulation of medical devices. The FDA’s Centre for Devices and Radiological Health (CDRH) is responsible for regulating companies that manufacture, repackage, relabel and/or import medical devices into the US. The TGA is responsible for registering medical products under the Australian Register of Therapeutic Goods (ARTG). Of the two, the Australian regulatory system is more comparable to the European system.
Whilst strict regulation may be seen as a trade-off against innovation, effective regulation can encourage and maintain innovation flows by providing assurance to consumers about the safety of the devices they use. I
Device Risk Levels
The US system uses three levels of risk classes. Unlike most medical regulatory authorities, it does not have two medium risk level classes. Under the FDA system, 43% of all devices are assigned to class II (source, FDA, 2017), making it arguably the most diverse class in terms of the range of risk profiles. The benefit of having three risk levels is simplicity: low-, medium- and high-risk categories.
The TGA uses the more commonly used system of four main classes (class I, class IIa, class IIb, class III), and additionally, a separate classification for Active Implantable Medical Devices (AIMD). Typically, class IIa devices can be evaluated using bench testing, whilst class IIb require some form of clinical testing.
This system is fairly consistent with markets in Europe, Japan and Canada. The TGA recently considered changing its classification and reclassifying all AIMDs and accessories as class III devices. This was met with backlash from the Medical Technology Association of Australia (MTAA), which said: “These changes would impose an unreasonable burden on manufacturers and sponsors, disproportionate to their intrinsic risk.”
The TGA, in response, decided to reclassify only surgically invasive and implantable accessories from the AIMD to the class III category of devices.
Medium-Risk Device Category
In comparing the two systems, it is evident that the biggest difference lies in the approach towards medium risk devices. One benefit of having a split risk level is that different controls can applied to the wide spectrum of devices that belong in these categories. Another benefit is that the system is harmonized with the majority of global regulatory systems.
On the other hand, the FDA still applies different controls (general and special controls) to devices that fall into the medium-risk level and therefore subjects higher risk devices to stricter regulation without the need for split risk levels. Furthermore, splitting the medium risk level may present an unnecessary burden to companies and hospitals, without delivering significant benefits.
Approval Pathways
Internationally, the approach to the regulation of medical devices involves evaluation of safety and effectiveness and/or efficacy, in order to secure approval for market launch. The approach differs from country to country, for historical and political reasons.
The two basic regulatory pathways for the FDA are the 510(k) and PMA pathway. The 510(k) route attracts significant controversy. In principle, it requires innovators to prove their (low-to medium-risk) devices are substantially equivalent to devices already on the market. Innovators justify their registrations on the premise of similar safety profiles. There is no requirement for clinical trials.
Historically, the 510(k) covered the thousands of devices that were already on the market when regulation was first introduced in the US. This route enabled newer versions of existing devices to enter the market quickly, without the burden of extensive clinical evaluation for novel devices.
Under a 510(k), manufacturers must prove their product has similar safety, effectiveness and indications for use, typically including a comparison of specifications, materials and technology used in the device. However, during the first Bush Administration, the 510(k) provision was amended to include products made from different materials and using different mechanisms of action. The rationale for doing so was to make the regulatory standard comply with the least burdensome approach.
US 510(k) Principles
The 510(k) pathway encourages innovators to make devices similar to those already available, since it is an easier method of regulatory approval.
In most cases, innovative devices are deemed as high-risk and must go through longer approval pathways. This reduces the incentive for innovation; products may only have slight iterationst from their competitors, as opposed from being radically different and ideally substantially better. The indications for use in the new device must be very similar to the predicate device to use this pathway, even though the newer device may have a broader range of uses.
Beyond the perceived innovation problems, the 510(k) system’s substantial equivalence is not seen as an effective form of regulation: the FDA is seen as evaluating differences between predicate devices and new devices to determine safety and effectiveness, rather than considering them independently.
However, for devices that are genuinely similar to devices already on the market where the patent has expired, the 510(k) process does provide a mechanism for fast approval. This benefits consumers by creating more competition and cheaper products. A recent example of this is seen in the FDA’s approval of the first generic Albuterol Inhaler to treat and prevent bronchospasm.
An Alternative To The 510(k)
A 2011 Institute of Medicine (IoM) committee recommended that the FDA eliminate the 510(k) rather than “continuing to modify a 35-year-old 510(k) process.” It pressed, instead, for more investment in an integrated pre-market and post-market regulatory framework that provides "reasonable assurance of safety and effectiveness throughout a product's lifecycle."
The US courts stated that “the 510(k) process is focused on equivalence, not safety.” To reform the system, the FDA should consider using accepted performance standards, rather than comparisons to predicate devices to demonstrate the safety of the innovation being cleared, it said. Furthermore, it was recommended that assessments should consider whether benefits to patient outcomes accrue from new devices that are superior to prior treatments.
Since these recommendations to eliminate the 510(k) process, the FDA has not changed the system but has placed more emphasis on the already existing de novo pathway.
De Novo Encourages Innovation
The de novo pathway provides a solution, as qualifying devices do not have to rely on being substantially equivalent to a legally-marketed predicate device. Instead, for novel medical devices that rely on general controls, there must be reasonable assurance of safety and effectiveness for the intended use.
This approach is effective at encouraging innovation, and of effective regulation, as companies must demonstrate strong safety and efficacy, as well as a robust risk management strategy.
“Post-amendments” devices were not in commercial distribution prior to 1976; they were automatically assigned to class III until a device was reclassified through the de novo process.
The statutory default of classifying novel products as class III products hampered innovation, as it created the need for reclassification. Furthermore, using reference products marketed before 1976 was not a suitable measure of performance, and, moreover, is outdated, according to David Feigal in 2010.
US pre-market approval (PMA) was intended to be used for all class III devices that were implantable or life-sustaining, or that presented significant risk to health safety or welfare of patient. The PMA allows the FDA to conduct inspections of manufacturing facilities to verify compliance with Quality System requirements. It has been criticized for being a slow process.
The US Government Accountability Office (GAO), in a 2009 review of the 510(k) process, found that 66% of class III devices cleared through the 510(k) process were implantables, life sustaining or of significant risk. This means they should have been required, by law, to go through the more stringent PMA process. However, despite these findings, the FDA has not changed its strategy.
In order to improve the FDA system, a possible solution from the IoM is to require that all lifesaving and life sustaining devices, such as cardiac implants, spinal implants and joint replacements, provide clinical trial evidence of safety and effectiveness.
The TGA regulates therapeutic goods through pre-market assessment, post market monitoring and enforcement of standards, licensing of Australian manufacturers and verifying that overseas manufacturers comply with standards.
Most sponsors choose to use overseas certification, such as EU certification, a Canadian license or US FDA approval, in support of their product’s inclusion in the ARTG. The manufacturer provides a technical file describing the product, intended purpose, risk classification and relevant standards. This is a risk-based approach that considers the intended use, duration of use and degree of invasiveness.
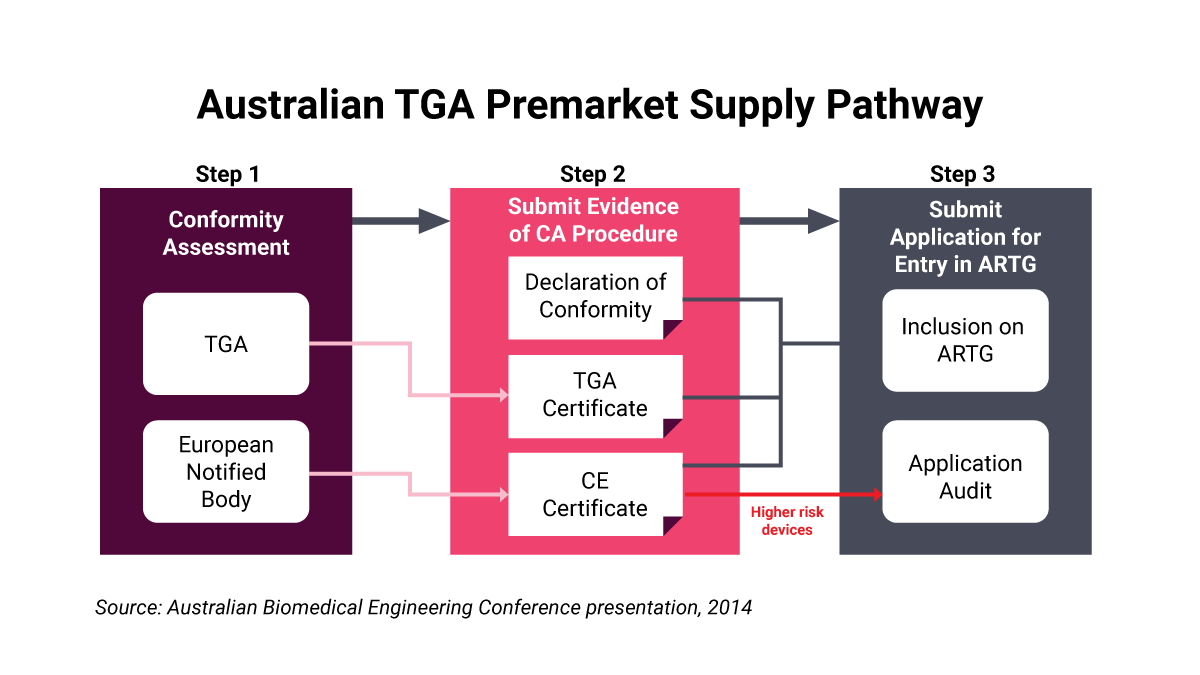
The figure (left) demonstrates the pre-market approval process and how the process may be faster if the company has already been given CE marking approval.
High-risk devices must hold a TGA-issued conformity assessment certification to be supplied in Australia. This means sponsors must provide information “on procedures used and evidence generated by the manufacturer to demonstrate that a medical device is designed and produced to be safe, fit for purpose and perform as intended.” Risk management documentation and clinical evidence supporting data should be included.
Unlike the FDA, at the TGA not all evaluation is done in-house, as it uses third party experts to assist with more specialized devices. The TGA has sought to improve the efficiency of its advisory committees, following the recommendations of the Medicines and Medical Devices Regulation Review Hub.The TGA's approval processes are consistent and transparent, and are seen as effectively balancing regulation and innovation needs.
Life Cycle Approach
The life cycle concept seeks to foster high quality, safety and efficacy of medical devices during the pre-market and post-market stages, and when medical devices have been implanted in patients for a lifetime.
The US 21st Century Cures Act, passed in 2016, is designed to accelerate medical product development to get innovations to patients quicker. The FDA has reformed its Q-Submission (Q-Sub) Program (which includes pre-submissions and other opportunities to engage with the agency) allowing for more dialog between regulators and innovators for their medical device submissions. This improves the quality of submissions, and shortens review times, since problems are remedied at an earlier stage.
The FDA recently implemented a voluntary program called STeP (Safer Technologies Program for Medical Devices) to expedite the development, assessment and review of devices. Its is expected to improve the safety of treatments and diagnostics available on the market. Sponsors can request to be included in STeP through a Q-submission. The benefits include prioritized review and senior management engagement. It encourages safer innovations that significantly improve the safety of devices already on the market.
The FDA allows manufacturers to make changes to devices cleared using the 510(k) system without notifying it, and does not require annual reporting. It is recommended that if manufacturers modify a device, they should notify the FDA of changes being made.
On the other hand, the Australian system has a large focus on post-market assessment.
Unlike other regulatory agencies, the FDA allows for public comment prior to publishing new regulation, allowing patients to give feedback on regulation reforms. This demonstrates that the FDA is reasonably effective at assisting innovators in getting their devices approved before commercialization.
FDA enforcement actions and post-market surveillance declined as a result of the Trump Administration’s deregulation efforts. A third fewer “warning letters” were sent to businesses, compared to the record of activity under the Obama Administration. Far fewer injunctions have been granted of late.
This means that defective products have a greater chance of remaining on the market, due to less strict post-market monitoring of adverse events and clinical data. However, the FDA has defended itseld, saying that “post-market vigilance actions are less discernible to the public, as the need for warning letters is reduced by meetings with companies, follow up inspections, amongst other behind-the-scenes action.”
The TGA places great emphasis on monitoring medical devices throughout their lifecycles. This includes considering how medical devices are designed, produced, supplied and disposed of. The approach relies on adverse incidents being reported by medical device users and sets mandatory reporting requirements for manufacturers.
But a reliance on user reporting can create problems, as doctors, nurses and biomedical engineers may be reluctant to fulfil the administrative requirements of reporting adverse events. Following an review, the TGA has decided it will disseminate information and/or oversee corrective action. The TGA exchanges information about medical devices incidents with other regulatory agencies.
The Australian system operates a patient registry for orthopedic devices, meaning that if a recall is necessary, a product can be traced back to the physician and patient. This effective form of regulation is intended to increase the efficiency of recalls, and considers the safety of patients. The possibility of a registry for orthopedic devices is in consideration in the US, however it is yet to be implemented.
Clinical Evidence Needs
The FDA has been collaborating with the Clinical Trials Transformative Initiative (CTTI) at Duke University, to rectify the increasing clinical trial costs and time spent addressing issues faced by innovators.
The US system requires a large number of test patients, compared to European and Australian regulatory requirements. The US has a strong preference for clinical trials conducted in the US when they are going through the PMA approval process.
Australian clinical trials are well respected around the world, due to its diverse population and high-quality research and facilities available. The TGA has a Clinical Trials Notification scheme that focuses on allowing for efficient ethics and scientific approval (usually within 4-8 weeks), which allows trials to start quicker.
The Australian government has introduced tax incentives to encourage research and development, particularly to encourage companies to conduct clinical trials in Australia. The TGA places a great emphasis on post-market data collection and monitoring the safety of the device while it is on the market.
Both Australian and US clinical trials do not require diversity in clinical trials to determine if new drugs or devices are efficacious in major demographic subgroups, such as Australia’s indigenous population or the US’s African American population. The FDA made recommendations in a recent guidance document to “enhance diversity in clinical trials populations” by broadening eligibility criteria and making trials less burdensome for study participants.
MDSAP And Global Collaboration
The US and Australian regulators have become part of the MDSAP, along with those from Canada, Japan and Brazil. This program allows a MDSAP-recognized auditor to conduct a single regulatory audit of a medical device manufacturer, in place of routine agency inspections conducted in each country. This increases efficiency, as businesses do not have to prepare for multiple audits with different processes. As a result, less time and money are spent on meeting regulatory requirements that can instead be put into innovating devices.
The US and Australia have adopted internationally recognized quality assurance standards, including ISO 9001 and ISO 13485. Standards promote a uniform regulatory framework, and the predictability they create enables innovations to be spread in markets globally.
The US has been criticized for lacking uniformity with rest of the world, as it did not assume some of the suggestions established by the Global Harmonization Task Force (GHTF) – now the International Medical Device Regulators Forum (IMDRF). The FDA has likely chosen not to conform in most areas due its reliance on its pre-established 510(k) system. The purpose of the GHTF was to establish consistency in global regulatory practices and promote technological innovation by improving efficiency. The GHTF has sought to harmonize premarket regulation, postmarket issues, quality-management systems, auditing and clinical evidence.
The TGA, along with the EU, Japan and Canada, has adopted the regulatory frameworks discussed in the IMDRF and precursor GHTF, since they were similar to the strategies being utilized already. Harmonization improves transparency, reduces regulatory burdens and promotes industry compliance. This increased efficiency allows innovations to enter the market quicker and easier.
The IMDRF is an example of how universal standards assist innovation by establishing uniform processes, rather than forcing companies to navigate complex and unique regulatory processes in each market they intend to enter. The TGA’s choice to actively engage in harmonization has allowed for more efficient and effective regulation and innovation.
Costs And Funding
The US in encourages innovation through its Small Business Determination (SBD) program, which allows start-ups to get products regulated by the FDA at lower cost. Small businesses with gross sales of less than $30m are eligible to have their first PMA waived.
This is important in encouraging innovation and competition, as it lowers the barrier for market entry, which can cause many companies to go bankrupt during the regulatory approval stage. Small and medium-sized businesses, more than big companies, are known to attempt to bring more novel medicines, devices and biologics to market.
The FDA has been effective at encouraging innovation by lowering the fees for SMEs to a quarter of the fees for large enterprises. The fees that the FDA charges for devices are: $11,594 for 510(k); $340,995 for a PMA; and $102,299 for the de novo process. Small businesses pay only 25% of these fees. These fees have been reformed under the Medical Device User Fee Amendments (MDUFA). They encourage effective regulation and perpetuate innovation, as most companies can afford the subsidised fees.
There is still an incentive to rely on the 510(k) process rather than the more expensive PMA. The FDA is largely government funded, which can create problems that affect regulation and innovation.
In contrast, the TGA is largely industry funded, with a small amount of government funding. Following the transvaginal mesh scandal, the TGA was criticized for having a conflict of interest. It rejected the claims, explaining that fees are used to fund staff time, regardless of the application being withdrawn, rejected or accepted.
The TGA has an annual charge exemption scheme, which allows for annual charges to be exempted until a product generates turnover. This is intended to encourage innovation, but it could be seen an incentive for the TGA to get products onto the market that are unsafe, in in order to collect an annual fee. There is little evidence that this is the case.
On the basis of the foregoing, the TGA funding system appears to achieve a better balance between regulation and innovation, as it does not create a bias towards less stringent approval processes due to cost constraints. As is also the case in the US, it does set lower fees for small and medium businesses to enable innovation.
Two Systems Compared
Medical device regulators must ensure that devices are safe and effective to use. At the same time, regulators should foster innovation in the medical devices. In some cases, stricter regulation can be to the detriment of innovation. It is the role of the TGA and FDA to balance the needs of industry with their role of protecting the public's health and safety.
Ensuring effective and standardized regulations promotes predictability in the process, and innovation can occur as a result. Both the FDA and TGA are taking steps to reform their processes to allow for a more streamlined approval processes that encourage innovation. The FDA is limited by the processes set up for the 510(k) route, which is open to criticicms for not being stringent enough and for limiting innovation, due to the requirement for devices to be similar to predicate devices.
The US system was established early, in global terms, and is therefore widely respected. It is a unique system, but it places a regulatory burden on businesses that would like to enter multiple markets.
Australia’s TGA balances regulation and innovation with a streamlined approach that is responsive to calls for reform aimed at improving efficiency and enhancing monitoring.
Claire Grimble is a member of the Asia Regulatory Professional Association (ARPA), and is affiliated to the NanYang Technological University, Singapore, and University of Technology, Sydney.