A Record-Low 24 Warning Letters Were Sent To Device Firms In 2018. That's Because A 'Big Hammer' Isn't The Best Tool To Ensure Compliance, FDA Says

Executive Summary
Only two dozen quality-related warning letters were sent by the US agency to device-makers in calendar year 2018 – the fewest ever issued to companies since the FDA's Quality System Regulation came into force in 1996. Two FDA officials share with Medtech Insight probable reasons for the decline in the enforcement missives, from regulatory meetings with firms to untitled letters, and more.
A mere two dozen quality-related warning letters were sent by the US Food and Drug Administration to device-makers in calendar year 2018 – the fewest ever issued to companies since the agency's Quality System Regulation came into force 23 years ago.
The 24 letters (17 domestic, seven foreign) mark a decrease of 31% from 2017, when 35 manufacturers received missives – which, at the time, was a record low. (Also see "FDA Sends Record-Low Warning Letters To Device-Makers In 2017 As Agency Takes More Personalized Compliance, Enforcement Tack" - Medtech Insight, 2 Apr, 2018.)
Meanwhile, Medtech Insight, through our Warning Letter Data Tracker, also counted 24 quality-related warning letters posted on the FDA's website in CY 2018.
The agency considers quality-related letters to be those that only include an alleged violation of the QSR. In its count, the agency does not include letters that solely pertain to the Medical Device Reporting (MDR) regulation (21 CFR, Part 803) or the Corrections and Removals regulation (21 CFR, Part 806), or premarket activities.
"Using the big hammer isn't necessary all the time." – Adam Saltman
The FDA's tabulation of 24 warning letters last calendar year practically mirrors its fiscal year 2018 count of 25 letters (19 domestic, six foreign), which were handed out between 1 October 2017 and 30 September 2018. The FY count of 25 is the product of 1,350 domestic and 557 foreign GMP inspections conducted during that time, according to the agency's FY 2020 budget request.
Letter Count On The Decline For Years
That only 24 letters were issued to firms in 2018 should come as no surprise to industry-watchers who have seen the number of letters steadily decline since 2015, when 121 were sent to manufacturers. (See chart below.)
In fact, there have been so few warning letters issued to device firms over the past few years that it's easy to forget that in 2012 – not long ago – the agency sent its most warning letters ever: 164.
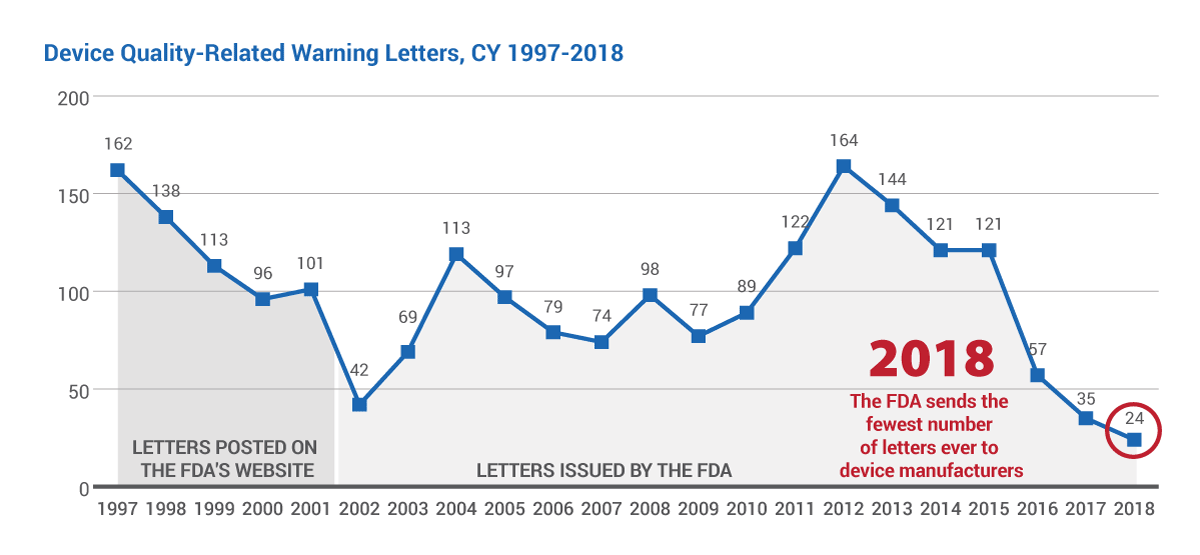 The FDA provided Medtech Insight with warning letter data for calendar years 2002-2018. Because the agency does not have an accurate count of its own for years 1997-2001, also provided in this chart is a tabulation of letters posted on FDA's website during that timeframe, as counted by Medtech Insight.
The FDA provided Medtech Insight with warning letter data for calendar years 2002-2018. Because the agency does not have an accurate count of its own for years 1997-2001, also provided in this chart is a tabulation of letters posted on FDA's website during that timeframe, as counted by Medtech Insight.
So why the sharp decline in letters?
Because "using the big hammer isn't necessary all the time," says Adam Saltman, a medical officer in the FDA device center's new Office of Product Evaluation and Quality (OPEQ).
At the Association of Food and Drug Officials (AFDO) annual conference in Atlanta, GA, on 25 June, Saltman told Medtech Insight that the agency considers benefit-risk factors and a firm's compliance history when determining whether to issue a warning letter.
"A lot of times there may an initial recommendation for a warning letter, but it ends up getting interpreted into an untitled letter or even a regulatory meeting to take care of the problem, the general idea being that a warning letter may not be necessary in all circumstances," he said.
Agency spokesperson Michael Felberbaum agreed with Saltman's assessment.
"The agency has a number of compliance and enforcement tools it can use to address quality system deficiencies or other issues with regulated industry, of which issuance of warning letters is one example," Felberbaum told Medtech Insight.
"The FDA commonly interacts with industry to resolve issues that often achieves more timely and effective corrective action to inspectional findings," he added. "For example, deficiencies may be resolved through alternative approaches such as holding regulatory meetings to discuss issues and expectations, or sending untitled letters to notify firms of violations and requesting follow-up action."
"We use a variety of tools to engage industry." – Michael Felberbaum
Felberbaum stressed that 2018's low warning letter count doesn't "account for other activities performed by the FDA to address inspectional findings, nor [does it] reflect the actions taken in follow-up to the inspection or the impact that inspections have on industry compliance with FDA requirements."
He pointed out that, between 2008 and 2017, 82% of firms corrected observed violations before their follow-up FDA inspection.
Further, Felberbaum said, "firms are eight times more likely to report a recall and reported more than three times the number of Medical Device Reports following an inspection over [that] same time period."
The FDA's popular-with-industry Case for Quality Voluntary Improvement Program (CFQ VIP) also likely plays a role in the reduction in warning letters, Felberbaum said.
CFQ VIP aims to elevate product, manufacturing and process quality at device firms by appraising the companies against an industry-modified version of the tried-and-true Capability Maturity Model Integration (CMMI) framework. (Also see "Chasing Quality Isn't Easy. But An FDA Pilot Aims To Boost Quality By Appraising The Capability Of Manufacturing Sites" - Medtech Insight, 7 May, 2018.)
"While issuing warning letters is an important tool for assuring compliance, our past experience is that device quality did not improve in response to issuing more warning letters," Felberbaum said. "Therefore, we use a variety of tools to engage industry and have explored faster approaches to effectively prevent or resolve issues, and improve industry actions to assure product quality."
Still, he said, warning letters are "an effective tool in achieving more timely and effective corrective action," noting that the agency would rather reserve the enforcement missives for "firms with severe violations" or those that "fail to implement or follow through on their corrective action plan in a timely fashion."